Genesis und das Genom
Das relativ neue und rasch wachsende Gebiet der vergleichenden Genomforschung liefert eine Fülle von Daten, die geeignet sind, um die Hypothese zu testen, dass Menschen und andere Lebensformen eine gemeinsame Abstammung haben. Zahlreiche unabhängige Ergebnisse der Genomforschung liefern starke Belege für die Hypothese, dass unsere Art einen gemeinsamen Vorfahren mit anderen Primaten hat. Weitere Hinweise zeigen auch, dass unsere Spezies bei ihrer Artbildung ausgehend von den Vorfahren der anderen Menschenaffen eine Populationsgröße von mindestens mehreren tausend Individuen hatte. Dieser Artikel gibt einen Überblick über Belege aus der Genomforschung für eine gemeinsame Abstammung und über Populationsgrößen der Hominiden. Außerdem werden die Konsequenzen dieser Indizienbeweise für naturwissenschaftlich-konkordistische Betrachtungsweisen der biblischen Schöpfungserzählungen kurz diskutiert.
Die Evolutionstheorie hat bereits früh vorgeschlagen, dass Menschen und andere Menschenaffen gemeinsame Vorfahren haben.1 Die Evolutionstheorie sagt damit voraus, dass die Genome, die wir in lebenden Primaten (wie Menschen und Schimpansen) finden, in Wirklichkeit modifizierte Formen eines ursprünglichen Genoms eines gemeinsamen Vorfahrens dieser Arten sind. Diese einfache Hypothese kann leicht mit Hilfe mehrerer unabhängiger Beweisführungen getestet werden, die sich aus dem Vergleich der vollständigen Genome der beiden Spezies ableiten.2
Die erste Beweisführung, und vielleicht die am meisten von christlichen apologetischen Organisationen diskutierte, ist die der Gensequenz-Ähnlichkeit. Wenn Menschen und Schimpansen tatsächlich von einer gemeinsamen Vorfahren-Spezies abstammen, dann würde man erwarten, dass die einzelnen Gen-Sequenzen dieser beiden Spezies einen hohen Grad an Ähnlichkeit aufweisen, weil sie von einem gemeinsamen Vorfahren vererbt wurden. Dies wird Homologie genannt. Darüber hinaus sollte die Homologie für einzelne Gene auf zwei Ebenen zu finden sein: der Aminosäure-Ebene (der funktionalen Sequenz des Protein-Produkts eines bestimmten Gens) und auf der Ebene des Nukleotid-Codes (des zugrunde liegenden DNA-Codes für die benötigte Aminosäuresequenz). Da der Nukleotid-Code zahlreiche Möglichkeiten für die Kodierung einer bestimmten Aminosäure-Sequenz bietet (d. h. der Nukleotid-Code ist redundant ), erwartet man, dass Gene in verwandten Organismen nicht nur gemeinsame Aminosäuresequenzen, sondern auch gemeinsame Nukleotidsequenzen haben, trotz einer großen Anzahl von möglichen Kodierungsmöglichkeiten. Folglich sollten verwandte Organismen Homologie auf beiden Code-Ebenen zeigen.
Eine zweite, unabhängige Beweisführung ist die der Syntenie. Syntenie ist ein Fachbegriff für die Erhaltung der Gen-Reihenfolge auf den Chromosomen von Verwandten. Vereinfacht gesagt: die Hypothese der gemeinsamen Abstammung sagt voraus, dass verwandte Arten nicht nur ähnliche Gene haben, sondern dass diese Gene in ihrer Anordnung auch ein sehr ähnliches räumliches Muster aufweisen.
Eine dritte Beweisführung ist die der Pseudogene. Pseudogene (wörtlich »unechte Gene«) sind die mutierten Reste von Gensequenzen, die in dem Genom nach ihrer Inaktivierung fortbestehen. Eine gemeinsame Abstammung lässt erwarten, dass verwandte Arten gemeinsame Pseudogene haben sollten, die in dem Genom ihres gemeinsamen Vorfahren vorhanden waren. Darüber hinaus sollten sich diese Pseudogene in beiden Nachkommenspezies an der gleichen Stelle im Genom befinden (d. h. sie sollten eine gemeinsame Syntenie zeigen) und trotz ihrer Inaktivierung eine Ähnlichkeit der Gensequenz erhalten haben (d. h. weiterhin Homologie zeigen).
Die DNA-Sequenzierung des menschlichen Genoms wurde zwischen 2001 und 2004 abgeschlossen und veröffentlicht.3 Kurz danach wurde die Sequenzierung des Schimpansen-Genoms abgeschlossen.4 Die Verfügbarkeit der vollständigen Genomsequenzen für beide Organismen ermöglicht einen Vergleich bezüglich Homologie, Syntenie und gemeinsamer Pseudogene über das gesamte Genom beider Spezies. Diese drei Analysen fungieren als unabhängige Tests für die Hypothese der gemeinsamen Abstammung von Mensch und Schimpanse und bieten damit voneinander unabhängige Belege in dieser wichtigen Frage.
Sequenzähnlichkeiten bei Primaten-Genen: Abstammungsnachweis durch Homologie
Homologie ist als Ähnlichkeit definiert, die sich von gemeinsamer Abstammung herleitet. Es ist seit langem bekannt, dass Menschen und Schimpansen für die einzelnen Gene nahezu identische Sequenzen besitzen.5 Die vollständige Genomsequenzierung hat bestätigt, dass dieses Schema der Beinahe-Identität über die Genome beider Arten hinweg zu finden ist. Das menschliche Genom hat etwa 3,0 x 109 Nukleotide; hiervon stimmen 2,7 x 109 Nukleotide bei nur 1,23% Abweichungen mit dem Schimpansen-Genom überein.6
Kurz gesagt, die große Mehrheit des menschlichen Genoms entspricht dem Genom des Schimpansen mit nur geringen Unterschieden. Die Einbeziehung von Sequenzalignment-Lücken zwischen den beiden Genomen, von denen man annimmt, dass sie entweder durch Insertionen oder Deletionen entstanden sind (sog. »Indels«), senkt die Identität der beiden Genome auf etwa 95% .7 Schränkt man den Vergleich auf die Sequenzen ein, die Proteine codieren, erhöht sich die Übereinstimmung auf 99,4%.8 Egal welches Maß man anlegt, Menschen und Schimpansen haben Genome, die hochgradig homolog sind und sich ohne Weiteres als modifizierte Kopien des Genoms eines ursprünglichen Vorfahren interpretieren lassen.
Codonverwendung in homologen Genen: Abstammungsnachweis durch Redundanz
Der DNA-Code, der benutzt wird, um Aminosäuren innerhalb von Proteinen festzulegen, basiert auf Nukleotidtripletts, auch »Codons« genannt. Da es vier Nukleotide (A, C, G und T) gibt, stehen 64 (d. h. 43) mögliche Nukleotidtripletts zur Verfügung; jedoch kommen nur zwanzig Aminosäuren in biologischen Proteinen vor. Da drei der 64 Codons als »Stopcodons« verwendet werden, um den Translationsprozess zu stoppen, stehen 61 Codons für die Kodierung der zwanzig Aminosäuren zur Verfügung. Deshalb können die meisten Aminosäuren durch mehr als ein Codon kodiert werden (d. h. der Codon-Code ist teilweise redundant). Beispielhaft wird ein Vergleich der Nukleotid- und Aminosäuresequenzen für Insulin (ein Peptidhormon) von Mensch, Schimpanse, Gorilla, Orang-Utan, einer Fledermausart und der Maus in Abbildung 1 gezeigt.9
Das Präproinsulinpeptid hat in allen sechs Spezies 110 Aminosäuren, von denen die meisten durch andere Codons kodiert werden können. Diese Redundanz im Code bedeutet, dass es über 1019 verschiedene mögliche Nukleotidsequenzen für Humaninsulin gibt, die ebenso die beobachtete Aminosäuresequenz ergeben. Die Sequenz, die wir tatsächlich vorfinden, ist jedoch nahezu identisch mit den Nukleotidsequenzen, die man in anderen Säugetieren sieht (Abbildung 1A). Die Schimpansen-Sequenz unterscheidet sich nur in sechs Nukleotiden; die des Gorilla nur in vier. Auf der Protein-Ebene unterscheiden sich Schimpansen in zwei Aminosäuren von den Menschen, während die Gorilla-Sequenz mit unserer identisch ist (Abbildung 1B). Die Aminosäuren- und Nukleotid-Homologien für andere Säugetiere unterscheiden sich in verschachtelter Weise zunehmend von der menschlichen Sequenz, und zwar genau übereinstimmend mit ihrer Phylogenie basierend auf morphologischen Kriterien (Abbildung 1C).
Obwohl dies eine sehr kleine Stichprobe ist (330 Nukleotide), ist dieses Muster repräsentativ: ein genomweiter Vergleich kodierender Sequenzen von Mensch und Schimpanse zeigt, dass sie bei 1,85 x 107 Nukleotiden zu 99,4% identisch sind.10
Abbildung 1. Nukleotid- und Aminosäure-Homologie für Insulin bei Säugetieren

Abbildung 1A. Die vollständige Nukleotid-Sequenz für Präproinsulin ausgerichtet (»aligned«) für vier Primatenarten (HS = Homo sapiens/Mensch, PT = Pan troglodytes/Schimpanse, GG = Gorilla gorilla/Gorilla, PP = Pongo pygmaeus/Borneo Orang-Utan), ein Chiroptera (RF = Rhinolophus ferrumequinum/Große Hufeisennase) und ein Murid (MM = Mus musculus/Maus). Nukleotide, die sich von der menschlichen Sequenz unterscheiden, sind schwarz unterlegt. Aminosäuren, die in allen sechs Spezies erhalten sind, sind unterhalb der Nukleotidsequenz angegeben. Die Zahlen unterhalb der in allen sechs Arten erhaltenen Codons geben die Anzahl der Codon-Alternativen für diese Position an.

Abbildung 1B. Die vollständige Aminosäuresequenz des Präproinsulin ausgerichtet für die gleichen Arten wie in (A). Aminosäuren, die sich von der menschlichen Sequenz unterscheiden, sind schwarz unterlegt.
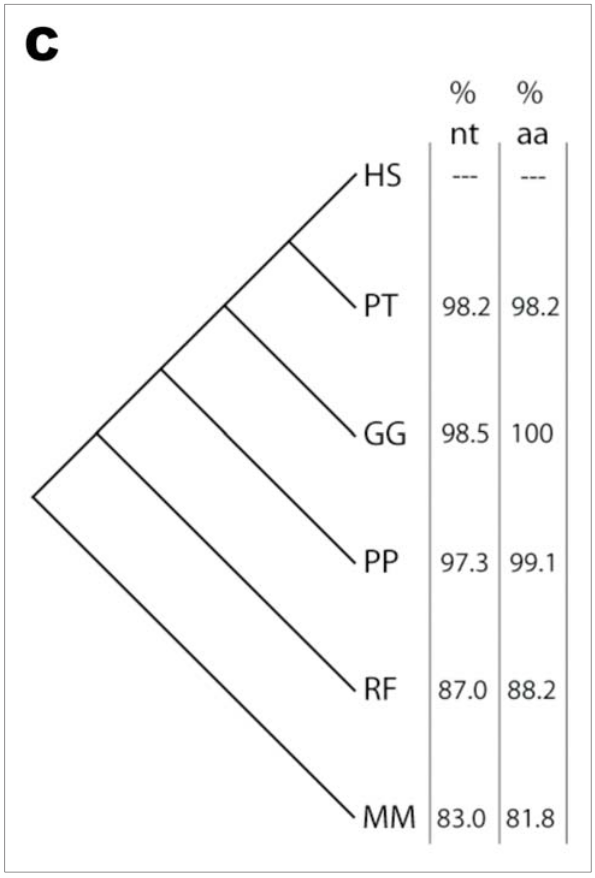
Abbildung 1C. Phylogenie für die gleichen sechs Arten, mit Prozentangaben für die Homologie bei Präproinsulin verglichen mit der menschlichen Sequenz für Nukleotid- (nt) und Aminosäure-Sequenzen (aa).
Dieses Argument kann auf Fälle ausgedehnt werden, in denen Unterschiede bei den Aminosäuren in bestimmten Proteinen zwischen verschiedenen Spezies beobachtet werden. Zum Beispiel sind die Unterschiede zwischen menschlichem und Schimpansen-Insulin auf der Ebene der Nukleinsäuren so klein wie möglich trotz verschiedener Aminosäuren. Die zwölfte Aminosäure im Schimpansen-Insulin beispielsweise ist Valin (Codon GTG), während es in den anderen hier untersuchten Säugetieren (Abbildungen 1A, 1B) Alanin (Codons GCG oder GCC) ist. Es gibt vier Codons, die Valin codieren (GT gefolgt von A, C, G oder T) und vier, die Alanin codieren (GC gefolgt von A, C, G oder T). Was wir sehen, wenn wir dieses Codon beim Menschen und Schimpansen vergleichen, sind die beiden ähnlichsten möglichen Codons trotz der veränderten Aminosäure. Oder anders ausgedrückt: der Nukleinsäure-Code passt zu einzelnen Nukleotidänderungen einer Sequenz eines gemeinsamen Vorfahren, obwohl es doch mehrere Möglichkeiten für die Codons der verschiedenen Aminosäuren gäbe.
Eine Ausweitung dieser Art der Analyse auf Insulin-Sequenzen von Organismen, von denen vorhergesagt wird, dass sie weniger eng mit den Menschen verwandt sind, bringt das gleiche Muster hervor: Gorillas und Orang-Utans verwenden das gleiche GCG-Codon für Alanin an der zwölften Position, während Fledermäuse und Mäuse ein GCC-Codon für dieses Alanin benutzen. Dieses Muster erstreckt sich über die gesamte codierende Sequenz für Insulin. Die signifikante Nukleinsäure-Homologie bleibt trotz der zahlreichen Möglichkeiten für die gleichbleibende Aminosäure-Sequenz erhalten (Abbildung 1C), und Änderungen stimmen bis auf Einzel-Nukleotid-Substitutionen sehr gut mit der Sequenz eines Vorfahren überein (Abbildung 1A). Als Zusammenfassung ergibt sich, dass die beobachteten Muster der Gen-Homologie über verschiedene Spezies hinweg auf zwei Code-Ebenen genau mit dem übereinstimmen, was eine gemeinsame Abstammung erwarten lässt.
Räumliche Organisation des Genoms: Abstammungsnachweis durch Syntenie
Syntenie, im Zusammenhang mit vergleichender Genomforschung, meint die Beobachtung, dass verwandte Organismen nicht nur hohe Sequenzhomologie für einzelne Gene besitzen, sondern dass auch die räumliche Organisation dieser Gene ähnlich ist. Kurz gesagt, Organismen, von denen man denkt, dass sie evolutionär nahe Verwandte sind, weisen im Wesentlichen die gleiche Gen-Reihenfolge auf – bis auf kleine Differenzen, die von bekannten Mechanismen wie Sequenz-Inversionen, Translokationen und Chromosomenfusionen stammen. Wie schon zuvor lässt die Hypothese der gemeinsamen Abstammung ein solches Ergebnis erwarten, da von den beiden betreffenden Spezies angenommen wird, dass sie einmal eine einzige Art waren.
Die Tatsache, dass die Genome von Mensch und Schimpanse auffallende Syntenie mit nur kleinsten Unterschieden in der genomischen Organisation zeigen, ist bereits seit einiger Zeit durch die Techniken der Chromosomenfärbung und der molekularen Hybridisierung bekannt.11 Die wichtigsten Unterschiede zwischen den Chromosomensätzen von Mensch und Schimpanse sind neun intrachromosomale Inversionen und eine Chromosomenfusion.12 Diese Beobachtungen wurden nun auf der molekularen Ebene durch Sequenzierung des gesamten Genoms von Mensch und Schimpanse bestätigt.13 Der vielleicht bekannteste Unterschied in der Genom-Organisation zwischen Menschen und Schimpansen ist die Telomer-an-Telomer-Fusion, aus der das menschliche Chromosom 2 entstand.14 Dieses Chromosom stimmt mit zwei getrennten Chromosomen bei Schimpansen und anderen Menschenaffen überein, was darauf hindeutet, dass das menschliche Chromosom das Ergebnis einer Fusion von zwei Chromosomen ist, die in diesen anderen Arten als zwei getrennte Chromosomen erhalten geblieben sind. Die Beweise für die Fusion beruhen auf Syntenie: die Gene der beiden Chromosomen der Affen sind genauso angeordnet wie in dem Menschenchromosom und entsprechen genau dem Muster, das man von einer Spitze-Spitze-Fusion erwarten würde.
Syntenie sagt auch voraus, wo bestimmte Nebenprodukte solch eines Fusionsereignisses zu erwarten sind. Chromosomen haben besondere Sequenzen an ihren Spitzen, die Telomere, sowie eine innere Sequenz, Zentromer genannt, die während der Zellteilung genutzt wird. Ausgehend von den beiden Chromosomen der Affen würde man vorhersagen, innere Telomer-Sequenzen genau dort im menschlichen Chromosom 2 zu finden, wo die Sequenzausrichtung mit dem einen Affenchromosom endet und die Sequenzübereinstimmung mit dem anderen beginnt. Wir würden auch die Anwesenheit von zwei Zentromeren vorhersagen, die genau in den Bereichen auftreten, wo sie in den Affenchromosomen gefunden werden. In beiden Fällen finden wir im menschlichen Chromosom 2 genau das, was bei einer gemeinsamen Abstammung zu erwarten wäre: interne Telomer-Sequenzen genau an dem vermuteten Fusionsort und das Vorhandensein von zwei Zentromeren an ihren vorhergesagten Positionen, von denen eines durch akkumulierte Mutationen inaktiviert wurde.15
Zusammenfassend lässt sich sagen, dass wir beim Vergleich der vollständigen Genome von Mensch und Schimpanse beobachten, dass die räumliche Organisation der Gene in beiden Arten genau dem entspricht, was man auf der Grundlage gemeinsamer Abstammung vorhersagen würde: überwältigende Ähnlichkeit mit kleinsten Unterschieden, die sich nach der Artbildung ergeben haben.
Genomische Archäologie: Abstammungsnachweis durch Pseudogene
Eine dritte und sehr überzeugende Beweisführung für gemeinsame Abstammung von Menschen und Menschenaffen ergibt sich vonseiten gemeinsamer Pseudogene. Pseudogene (wörtlich: »unechte Gene«) sind Gensequenzen, die durch Mutationen inaktiviert wurden und als nicht funktionsfähige Sequenzen im Genom fortbestehen. Pseudogene bleiben aus mehreren Gründen erkennbar. Zunächst sind nur kleine Änderungen notwendig, um ein Gen zu inaktivieren (zum Beispiel die Veränderung eines Codons zu einem störenden Stop-Codon, das die Protein-Translation abbricht). In solchen Fällen sind die Gen-»Reste« nahezu identisch mit dem funktionsfähigen Gen und durch ihre Homologie leicht erkennbar. Zweitens ermöglicht die vergleichende Genomforschung, Pseudogene nicht nur aufgrund von Sequenz-Homologie zu funktionsfähigen Genen in anderen Organismen zu identifizieren, sondern auch durch Syntenie: Pseudogene behalten ihre räumliche Orientierung zu benachbarten funktionsfähigen Genen nach ihrer Inaktivierung bei. Drittens akkumulieren Pseudogene, einmal inaktiviert, Mutationen nur langsam, weil die Korrekturlesemechanismen, die die DNA-Replikation steuern, nicht zwischen funktionsfähigen und nichtfunktionsfähigen DNA-Sequenzen unterscheiden. Diese Merkmale ermöglichen eine Identifikation der Pseudogene in verschiedenen Stadien des Verfalls, während sie langsam über Millionen von Generationen bis zur Unkenntlichkeit mutieren.16
Gemeinsame Abstammung lässt auch erwarten, dass, jenseits des gemeinsamen Mensch-Schimpanse-Vorfahren, der gemeinsame Primaten-Vorfahre in der ferneren Vergangenheit auch einen gemeinsamen Vorfahren mit anderen Wirbeltieren hat. Zum Beispiel sagt die Evolutionstheorie voraus, dass die Menschen, wie alle Wirbeltiere, von eierlegenden Vorfahren abstammen.17 Wie alle Plazenta-Säugetiere verwenden Menschen nicht Ei-Dotter als Nahrungsquelle für ihre Embryonen. Andere Wirbeltiere wie Fische und Vögel nutzen Ei-Dotter, wie auch eine kleine Anzahl noch vorhandener Säugetierarten wie das Schnabeltier.
Ein Protein, das als Dotter-Komponente in eierlegenden Wirbeltieren vorkommt, ist das Produkt des Vitellogenin-Gens.18 Da man annimmt, dass plazentale Säugetiere von eierlegenden Vorfahren abstammen, haben Forscher vor kurzem untersucht, ob Menschen Reste der Vitellogenin-Gen-Sequenz als Pseudogen zurückbehalten haben. Um die Suche zu erleichtern, bestimmte diese Gruppe die Position des funktionsfähigen Vitellogenin-Gens im Hühner-Genom, stellte die Identität der Gene fest, die an die Vitellogenin-Sequenz angrenzen und lokalisierte diese Gene im menschlichen Genom. Die Forscher fanden, dass diese Gene im menschlichen Genom ebenfalls nebeneinander vorhanden und funktionsfähig sind; dann untersuchten sie die Sequenz im menschlichen Genom zwischen ihnen. Wie erwartet, war die stark mutierte, pseudogenisierte Sequenz des Vitellogenin-Gens im menschlichen Genom genau an dieser Position vorhanden.19 Das menschliche Genom enthält also die mutierten Reste eines Gens, das in eierlegenden Wirbeltieren der Eidotter-Bildung dient, genau an der Stelle, die aufgrund von Syntenie bei einer gemeinsamen Abstammung vorhergesagt wurde.
So überzeugend das Vitellogenin-Pseudogen auch ist, ist es nur ein Beispiel von Tausenden, die präsentiert werden könnten.20 Beispielsweise gibt es Hunderte von Genen im menschlichen Genom, die für den Geruchssinn zuständig sind (olfaktorische Rezeptor-Gene), die aber zu Pseudogenen geworden sind.21 Viele dieser Pseudogene weisen bei Menschen, Schimpansen und Gorillas identische inaktivierende Mutationen auf.22 Wenn man den Verwandtschaftsgrad allein anhand von identischen inaktivierenden Mutationen in Geruchsrezeptor-Pseudogenen bestimmen wollte, so ergibt sich, dass der Mensch am engsten mit Schimpansen verwandt ist (höchste Anzahl gemeinsamer Fehler), dann als nächstes mit Gorillas (weniger gemeinsame Fehler) und dann mit Orang-Utans (noch weniger gemeinsame Fehler).23 Ferner wurden in der Studie auch keine Pseudogene gefunden, die aus der Reihe tanzen würden: Pseudogene mit identischen inaktivierenden Mutationen, die in Menschen und Gorillas gemeinsam auftreten, waren mit den identischen Mutationen auch bei Schimpansen vorhanden; Mutationen die bei Menschen und Orang-Utans gemeinsam auftreten, waren mit den identischen Mutationen auch in Schimpansen und Gorillas vorhanden.
Dieses Muster ist genau das, was gemeinsame Abstammung für diese Spezies erwarten lässt, da eine identische Mutation in beiden Arten am natürlichsten dadurch erklärt werden kann, dass sie bereits bei einem gemeinsamen Vorfahren beider Spezies vorhanden war. Der gemeinsame Vorfahre von Mensch und Gorilla ist auch der gemeinsame Vorfahre der Schimpansen. Demzufolge erwartet man inaktivierende Mutationen bei Menschen und Gorillas in gleicher Weise dann auch bei Schimpansen. Kurz gesagt, das Vorhandensein gemeinsamer Pseudogene in Primaten-Genomen, ihre syntenische Position und das Muster ihrer Inaktivierung und Verteilung unterstützen alle einheitlich das gleiche Modell der gemeinsamen Abstammung, das bereits allein auf Basis der Kriterien der vergleichenden Sequenz-Homologie aufgestellt wurde.
Vergleichende Genomforschung: Abstammungsnachweise oder gemeinsames Design
Während Belege aus der Genomforschung in Form von Homologie, Syntenie und Pseudogenen unabhängig voneinander die Hypothese unterstützen, dass Menschen und Schimpansen einen gemeinsamen Vorfahren haben, ist es auch möglich, diese Indizien aus einer Sicht zu bewerten, die nicht von gemeinsamer Abstammung ausgeht, wie beispielsweise dem Konzept des Intelligent Design (ID). Obwohl es stimmt, dass einige Wenige innerhalb der ID-Bewegung eine gemeinsame Abstammung von Mensch und Schimpanse akzeptieren,24 scheint diese Position aber doch eine Minderheitenmeinung in der Gruppierung zu sein, die eine Erklärung durch gemeinsames Design anstelle einer gemeinsamen Abstammung bevorzugt.25 Während eine vollständige Behandlung dieses Themas den Rahmen dieses Artikels sprengen würde, ist ein kurzer Überblick über die Einordnung der Erkenntnisse aus der Genomforschung in einem Rahmenkonzept, das gemeinsame Abstammung leugnet, lehrreich für die Untersuchung der relativen Stärken und Schwächen von ID einerseits und der Standard-Evolutionsanschauung, die auf gemeinsamer Abstammung basiert, andererseits in ihrem Bestreben, die Daten der vergleichenden Genomforschung bei Primaten zu erklären.
Homologie, Redundanz und Gemeinsames Design
»Warum sollte der Designer nicht auch ähnliche DNA und ähnlichen Körperbau für verschiedene Organismen verwenden? Genetische Ähnlichkeit zwischen Schimpansen und Menschen macht von einem evolutionären Standpunkt aus Sinn, aber sie steht auch im Einklang mit intelligentem Design.« 26
»[...] Designer verwenden oft einmal entworfene Teilkonstruktionen für unterschiedliche Anwendungen wieder. Wenn ein Designer eine menschenähnliche Spezies erzeugen wollte, so folgt natürlich, dass er viele gleiche Gene wiederverwenden würde.« 27
Es ist vielleicht vernünftig zu schlussfolgern, dass ein Designer einmal entworfene Teile wiederverwenden könnte, wenn er ähnliche Konstruktionen durch jeweils spezielle Schöpfung umsetzen möchte. Jedoch beobachtet man, dass Menschen- und Schimpansen-Gene nicht nur auf der Ebene der Aminosäuren (d. h. der funktionalen Ebene), sondern auch in deren zugrunde liegenden Nukleotid-Codes übereinstimmen.
Wie wir oben gesehen haben, gibt es eine Vielzahl von Nukleotid-Sequenzen, die einem Designer zur Verfügung stehen, um eine gegebene Aminosäuresequenz zu kodieren. Selbst wenn ein Designer gezwungen wäre, die gleiche Aminosäuresequenz zu benutzen, um gleiche Protein-Funktionalität in ähnlichen Organismen zu erreichen (was an sich bereits fraglich ist, da nicht-homologe Enzyme die gleiche Reaktion durchführen können), wäre es für einen solchen Designer ein Leichtes, einen wechselnden Nukleotid-Code zu wählen, um den Anschein einer gemeinsamen Abstammung zu vermeiden. Doch was man immer wieder beobachtet ist, dass genetische Codes in Organismen, von denen man aufgrund nicht-genetischer Kriterien annimmt, dass sie nahe evolutionäre Verwandte sind, sowohl auf den Nukleotid- als auch auf den Aminosäure-Ebenen übereinstimmen. Das ist genau das, was gemeinsame Abstammung erwarten lässt, da die Hypothese die ist, dass ähnliche Organismen einst die gleiche Spezies mit identischen Genomen waren. Einer Design-Auffassung, die gemeinsame Abstammung leugnet, macht dieses Muster Probleme. Es deutet darauf hin, dass der Designer den überwältigenden Anschein einer gemeinsamen Abstammung nicht vermeiden wollte (oder noch schlimmer, dazu nicht in der Lage war), als er seine Konstruktionen in vermeintlich getrennt geschaffenen Organismen umsetzte.
Syntenie und gemeinsames Design
In der ID-Literatur wird Syntenie selten diskutiert, und wenn, dann nur sehr schlecht begründet. Es wird beispielsweise versucht, die Schlussfolgerung zu entkräften, dass die Zeichen der Chromosomen-Fusion im menschlichen Chromosom 2 eine gemeinsame Abstammung untermauern. Hieran werden die wesentlichen Argumente erkennbar:
»[...] die Indizien für die Chromosomen-Fusion stärken einfach nur die Hinweise auf genetische Ähnlichkeit zwischen Schimpansen und Menschen. Da Ähnlichkeit auch ohne Darwinismus und gemeinsamer Abstammung erwartet werden konnte, können Ähnlichkeiten zwischen Organismen genauso gut das Ergebnis von funktionellen Anforderungen sein, die durch ein gemeinsames Design umgesetzt wurden.« 28
Dieses Argument entzieht sich, wie wir gesehen haben, dem Sachverhalt, dass Syntenie und Homologie von dem Standpunkt eines gemeinsamen Designs nicht zwangsläufig zusammen zu erwarten sind.
Darüber hinaus wird in der ID-Literatur nicht erwähnt, dass diese vorhergesagte »Erfordernis gemeinsamer Syntenie« gerade nicht gefunden wird, wenn die Genome von anderen Gruppen sehr ähnlicher Organismen verglichen werden. Beispielsweise liegen jetzt die vollständigen Genomsequenzen von zwölf Fruchtfliegen-Arten (Drosophila) vor29 und ihre Genom-Organisationen wurden verglichen.30 Die Ergebnisse dieser Analysen zeigen, dass dem Körperbau und der Biochemie von Drosophila durch ein breites Spektrum von syntenischen Anordnungen genüge getan wird, wobei in dieser Gruppe deutlich vielfältigere chromosomale Umlagerungen beobachtet werden als zwischen Menschen und Schimpansen. Außerdem nimmt die Größe der syntenischen Gen-Blöcke zwischen den Drosophila-Arten mit zunehmendem zeitlichen Abstand von ihrer Artbildung, die mit molekularen Uhren ermittelt wurde, ab. Je unterschiedlicher die einzelnen Gen-Sequenzen zwischen zwei Arten sind, desto weniger Gene sind in syntenischen Gruppen erhalten.31 Einfacher ausgedrückt: der Designer scheint für Fruchtfliegen ein breiteres Spektrum von unterschiedlichen genomischen Organisationen eingesetzt zu haben, die alle die richtige biologische Funktion und Drosophila-Morphologie verleihen. Das Muster der abnehmenden Syntenie entspricht dem Muster der abnehmenden Gensequenz-Homologie, genau wie es eine gemeinsame Abstammung erwarten lässt. Obwohl die betreffenden Fliegenarten für einen Nicht-Spezialisten schwer mit dem Auge zu unterscheiden sind, sind die genetischen Unterschiede so groß, dass es leichter wäre zu argumentieren, dass verschiedene Drosophila-Arten getrennte, unabhängige Designs sind, als es ist zu argumentieren, dass Menschen und Schimpansen getrennte, unabhängige Designs sind.
Das Problem der ID-Argumentationsweise ähnelt dem, was wir bei der Redundanz gesehen haben. Es gibt, ausgehend von einem Design-Standpunkt, der gemeinsame Abstammung leugnet, a priori keinen Grund, ein Muster von ähnlicher genomischer Organisation (d. h. gemeinsamer Syntenie) für Menschen und Schimpansen zu erwarten. Es gäbe allen Grund ein sehr unterschiedliches Muster zu erwarten, das auf unabhängige spezielle Schöpfung hinwiese. Noch einmal: Der Syntenie-Nachweis untermauert nicht nur nachdrücklich die gemeinsame Abstammung von Mensch und Schimpanse, sondern ist auch höchst problematisch für Interpretationen, die eine gemeinsame Abstammung leugnen.
Pseudogene und gemeinsames Design
ID-Literatur, die gemeinsame Abstammung leugnet, stimmt bei der Behandlung von Pseudogenen in drei Merkmalen überein: (1) Zusammenfassung von Pseudogenen und aller nicht-kodierenden DNA in die Kategorie »Junk-DNA« (»Müll-DNA«); (2) keine Diskussion der Beobachtung, dass Pseudogene mit identischen inaktivierenden Mutationen bei verschiedenen Organismen in genau dem Muster vorkommen, wie es durch gemeinsame Abstammung erwartet wird; und (3) dem Vorschlag, dass Pseudogene bis jetzt noch ungeklärte Funktionen haben, die ihr Vorhandensein als Ergebnis bewussten Designs erklären.32 Das Argument der unbestimmten Pseudogen-Funktionen lässt die vielen Fälle unerwähnt, in denen eine Funktion für ein bestimmtes Genprodukt bekannt ist (und gerade dadurch eine Sequenz als Pseudogen identifiziert wurde). Zum Beispiel ist die Funktion des Vitellogenin-Gens in eierlegenden Lebewesen bekannt, ebenso wie die Funktion der zahlreichen Geruchsrezeptoren, die wir als Pseudogene beim Menschen und anderen Primaten finden.
Darüber hinaus geht die ID-Literatur nicht darauf ein, dass wir diese Pseudogene in genau der syntenischen Anordnung finden, die durch gemeinsame Abstammung vorhergesagt wird. Wenn man das Intelligent-Design-Argument annehmen möchte, muss man behaupten, dass der Designer diese Sequenzen an genau den syntenischen Stellen in das menschliche Genom platziert hat, wo sich in anderen Organismen funktionsfähige Versionen dieser Gene befinden. Und nicht nur das. Der Designer hätte auch in hohem Maße homologe Sequenzen benutzt, die offenkundig in abgestufter Folge gleiche Mutationen aufweisen, die genau zu den (davon unabhängigen) phylogenetischen Stammbäumen passen, um eine in keinem Zusammenhang damit stehende, bislang unbekannte Funktion zu erfüllen. Während eine solche Möglichkeit natürlich nie hundertprozentig ausgeschlossen werden kann, fragt man sich doch, warum der Designer eine Design-Methode wählen würde, die einen so überwältigenden Eindruck von gemeinsamer Abstammung der Lebewesen erzeugt.
Gemeinsames Design: eine Theorie in der Krise
Zusammenfassend lässt sich sagen, dass Homologie, Redundanz, Syntenie und gemeinsame Pseudogene unabhängige Beweisführungen der Genomforschung sind, die in eine einzige Schlussfolgerung münden: Menschen sind nicht biologisch unabhängige De-novo-Schöpfungen, sondern besitzen gemeinsame Vorfahren mit anderen Lebensformen. Darüber hinaus scheinen Bestrebungen, die Belege der Genomforschung von einem Intelligent-Design-Standpunkt aus zu deuten, der eine gemeinsame Abstammung verwirft und von einem gemeinsamen Design ausgeht, überaus erzwungen und äußerst ad hoc. Während jede Beweislinie der Genomforschung für einen ID-Standpunkt, der gemeinsame Abstammung leugnet, schon einzeln problematisch ist, sind die Belege in ihrer Kombination, die ein derart geschlossenes Bild ergibt, geradezu vernichtend.
Genomforschung und Populationsgrößen von Hominiden-Vorfahren: Die Frage nach Adam und Eva
Während sich viel Aufmerksamkeit auf die Folgerungen aus dem Humangenomprojekt hinsichtlich der gemeinsamen Abstammung mit anderen Primaten gerichtet hat, ermöglichen andere Fortschritte in der vergleichenden menschlichen Genomforschung Einblicke in andere Aspekte unserer biologischen Vergangenheit. Ein solcher Bereich ist die Anwendung der Kenntnisse über die genetische Variation des heutigen Menschen, um effektive Populationsgrößen unserer menschlichen Vorfahren zu verschiedenen Zeitpunkten unserer evolutionären Vergangenheit abzuschätzen.
Das Verfahren zur Schätzung von Populationsgrößen aus den Daten der vergleichenden Genomforschung ist quantitativer Natur33 und recht komplex, so dass es sich Laien nur schwer erschließt. Aber die Ergebnisse dieser Berechnungen sind sowohl qualitativ als auch quantitativ durchaus nachzuvollziehen. Zum Beispiel ist ein kleiner, aber signifikanter Anteil des menschlichen Genoms dem Genom des modernen Gorillas ähnlicher als dem Genom des Schimpansen.34 Für diese Untermenge von Sequenzen stimmt unser Speziesbaum nicht mit dem Genbaum überein (Abb. 2).35 Eine solche Diskrepanz wird für eng verwandte Arten, die sich erst vor kurzer Zeit voneinander getrennt haben, aber sogar erwartet.36 Anders ausgedrückt: Der Grund, weshalb unser menschliches Genom dem Schimpansen-Genom überhaupt so überwältigend viel ähnlicher ist als dem Gorilla-Genom, ist, dass wir vor nicht allzu langer Zeit einen gemeinsamen Vorfahren mit den Schimpansen hatten. Doch ungeachtet dessen haben wir offensichtlich einige Regionen in unserem Genom bewahrt, die enger mit Gorillas verwandt sind. Dies geschah, weil die Population, die den gemeinsamen Vorfahren von Mensch und Schimpanse hervorbrachte, groß genug und genetisch vielfältig genug war, um diese Variation an uns zu vererben, ohne sie auch an die Schimpansen zu vererben. Schimpansen und Menschen sind somit getrennte genomische Auswahlen aus einer vielfältigen Vorfahren-Population. Wäre dieser Genpool klein gewesen, würden die Genbäume von Mensch und Schimpanse in fast jedem Fall mit dem Speziesbaum übereinstimmen. Der Anteil der Genbäume, die nicht mit dem Speziesbaum übereinstimmt, kann somit zur Abschätzung der Populationsgröße der Vorfahren-Population benutzt werden.37
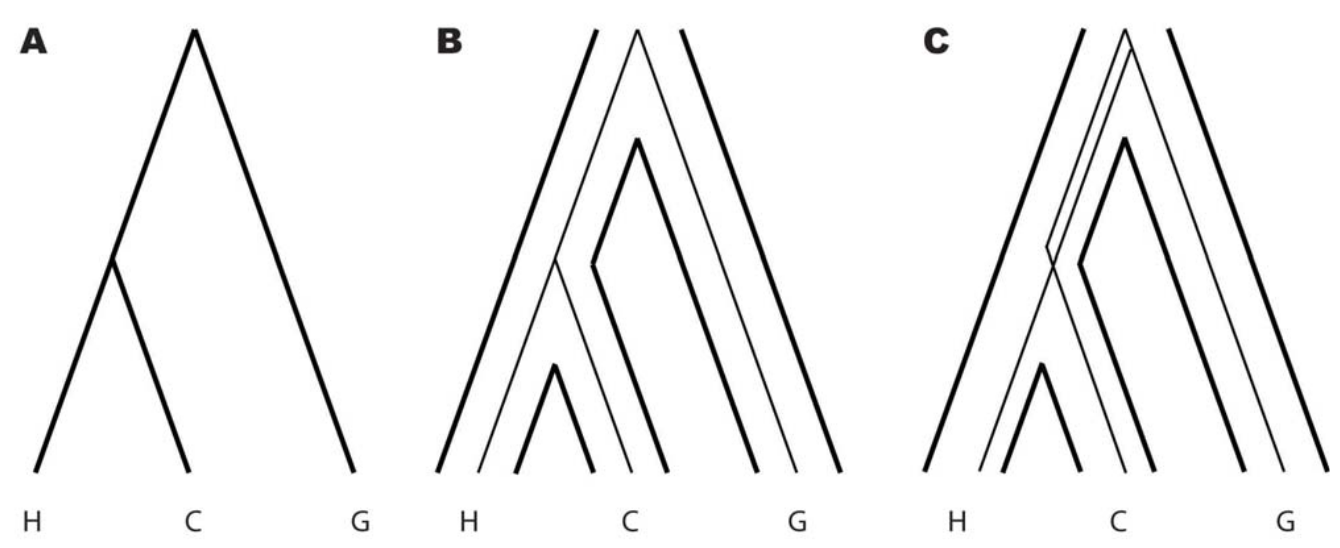
Abbildung 2. Spezies- und Genbäume für Mensch, Schimpanse und Gorilla
A. Die vergleichende Primaten-Genomforschung stützt nachdrücklich einen Primaten-Speziesbaum, der Menschen (H) und Schimpansen (C) so anordnet, dass ihre Trennung in jüngerer Zeit geschah als die Trennung von den Gorillas (G).
B. Die meisten Gene in Menschen und Schimpansen sind im Genbaum verbunden, bevor sie sich mit denen des Gorillas verbinden.
C. Eine Minderheit der Gene jedoch ist jeweils direkt mit Gorilla-Genen verbunden. Ein solcher alternativer Genbaum entsteht, wenn Varianten dieser Gene in der gemeinsamen Vorfahren-Population von Mensch und Schimpanse erhalten geblieben sind, nachdem die Gorillas sich abgetrennt haben. Dementsprechend kann der Anteil der Gene beim Menschen, deren Genbaum dem Speziesbaum widerspricht, verwendet werden, um auf die effektive Populationsgröße der Abstammungslinie zu schließen, die den Menschen der Gegenwart mit dem Punkt der Trennung vom Gorilla verbindet. Nähere Details im Text.
Frühe Studien, die nur mit eingeschränkten Datenmengen arbeiten konnten, schätzten übereinstimmend, dass die effektive Populationsgröße der Vorfahren von Homo sapiens im Bereich von 10.000 Individuen lag, wobei die Untergrenze des 90%-Konfidenzintervalls im Bereich von 6.000 liegt.38 Weil dieser Wert Schimpansen und/oder Gorillas als Vergleich verwendet, ist er ein Maß für die effektive Populationsgröße unserer Abstammungslinie seit der Artbildung der Schimpansen (vor ca. 4-6 Millionen Jahren) oder der Gorillas (vor ca. 6-9 Millionen Jahren).39 Sowohl die Verfügbarkeit des vollständig sequenzierten Schimpansen-Genoms als auch umfangreicher Sequenzen, die aus dem laufenden Gorilla-Genomprojekt zur Verfügung stehen, haben die Abschätzung dieser Vorfahren-Populationen mit zunehmender Präzision möglich gemacht. Übereinstimmend mit den älteren Studien kommen neuere Untersuchungen, die sehr große Datenmengen nutzen konnten, auf Schätzungen im Bereich von 8.000-10.000 Individuen.40
Die bis heute vielleicht anspruchsvollste Studie verwendet die inzwischen vollständigen Genomsequenzen von Mensch und Schimpanse, um alternative Genbäume für Sequenzen in situ innerhalb ihres menschlichen Chromosomenkontextes auszuwerten (d. h. unter Einbeziehung von Syntenie).41 Während diese Studie mit den bisherigen Schätzungen übereinstimmt, zeigt sie auch, dass die Sequenzen mit alternativem Baum (d. h. Sequenzen von Mensch und Gorilla laufen vor Mensch und Schimpanse zusammen) wie erwartet in kleinen Syntenie-Blöcken zusammen gruppiert sind.42
Fortschritte bei der Erforschung der genetischen Vielfalt innerhalb unserer Spezies haben ergänzende Methoden hervorgebracht, mit denen die effektive Populationsgröße unserer Vorfahren abgeschätzt werden kann. Die Annahmen, die diesen Methoden zugrunde liegen, sind unabhängig von jenen, die für Spezies-überschreitende Genom-vergleichende Ansätze angenommen werden. Das groß angelegte internationale HapMap-Projekt kartiert und katalogisiert menschliche Einzelnukleotid-Polymorphismen (SNPs, engl. single nucleotide polymorphisms; sprich: Snips).43 Während SNPs einzeln betrachtet eine Quelle genetischer Variation wie jede andere sind, können sie, wenn man sie als auf demselben Chromosom liegende gekoppelte Gruppen untersucht, genutzt werden, um die Populationsdynamik der Vorfahren mittels eines Effekts namens Kopplungsungleichgewicht (LD, engl. Linkage Disequilibrium) abzuschätzen.44
Weit entfernt liegende SNPs rekombinieren leicht während der Meiose, aber eng gekoppelte SNPs tun dies nicht, und sie neigen daher dazu, zusammen vererbt zu werden. Vergleicht man die Häufigkeit einzelner SNP-Allele mit ihren Kopplungsmustern zu anderen SNPs in der gleichen Population, zeigt sich, dass viele SNP-Paare im Kopplungsungleichgewicht (LD) sind: sie erweisen sich häufiger an andere SNP-Allele gekoppelt, als man aufgrund einer Zufallsverteilung erwarten würde. Die biologische Grundlage für LD ist, dass SNP-Paare von Vorfahren vererbt werden und sich innerhalb einer Population ausbreiten ohne aufgebrochen zu werden; eng zusammenliegende SNPs bleiben länger zusammen und weiter getrennte rekombinieren mit einer höheren Rate. Folglich können bekannte Rekombinationsraten zwischen SNPs und die Verteilung und Verhältnisse von SNP-Paaren in einer Population genutzt werden, um die Populationsgröße abzuschätzen.45 Da die Rekombinationsfrequenz durch die physikalische Distanz zwischen SNP-Paaren bestimmt ist, können LD-Studien genutzt werden, um die zeitliche Entwicklung von Populationsgrößen zu schätzen, was mutationsbasierte Schätzungen so nicht können. Die Auswahl von eng gekoppelten Markern ermöglicht Schätzungen in der ferneren Vergangenheit, während weiter entfernt liegende SNPs (mit ihren entsprechend höheren Rekombinationsraten) für Schätzungen in der jüngeren Vergangenheit dienen. Weil es außerdem viele Tausende von SNP-Paaren zum Untersuchen im menschlichen Genom gibt, liefert jede menschliche Populationsstichprobe eine Vielzahl von Datenpunkten für LD-basierte Methoden.
Auf SNP/LD-Ansätzen basierende Studien haben nun die zeitliche Dynamik der Vorfahrenpopulationen verschiedener Bevölkerungsgruppen detaillierter geschätzt, als dies mit mutationsbasierten Schätzungen möglich ist. Über die letzten 200.000 Jahre hinweg haben afrikanische Gruppen generell eine höhere effektive Populationsgröße (~ 7.000) als nicht-afrikanische Gruppen (~ 3.000) gehabt.46 Bemerkenswert ist, dass diese Studien damit die Schlussfolgerung älterer Studien untermauern, dass die Menschheit als Spezies von einer Vorfahrenpopulation mit mindestens einigen tausend Individuen abstammt – und das, obwohl dieser Ansatz auf Methoden und Annahmen beruht, die komplett unabhängig sind von früheren Studien. Noch wichtiger ist, dass durch die Skalierbarkeit dieses Ansatzes gezeigt werden konnte, dass es keine signifikante Veränderung in der menschlichen Populationsgröße gab: weder zu der Zeit, als die modernen Menschen im Fossilienbefund erscheinen (vor ~ 200.000 Jahren) noch in der Zeit bedeutender kultureller und religiöser Entwicklung vor ~ 50.000 Jahren.47
Sowohl einzeln als auch zusammen betrachtet, legen Studien der Populationsgenetik eindringlich nahe, dass unsere Abstammungslinie in den letzten 9.000.000 Jahren oder mehr keinen außergewöhnlichen Flaschenhals durchlebt hat (und damit weder in einer Hominiden- noch in einer Australopithecus-Art), ferner dass jegliche Flaschenhalseffekte, die unsere Abstammungslinie erfahren hat, die Population jeweils auf nicht weniger als einige Tausend sich fortpflanzende Individuen reduzierte. Von daher findet die Hypothese, dass die Menschen genetisch von einem einzigen Vorfahrenpaar in jüngster Vergangenheit abstammen, nicht die geringste Unterstützung aus der Genetik und widerspricht explizit einer enormen Menge von gegenteiligen Belegen.
Was aber ist mit der mitochondrialen Eva und dem Y-Chromosom-Adam?
Die oben präsentierten genomischen Daten können den Anschein erwecken, als stünden sie im Widerspruch zu der Beobachtung, dass menschliche mitochondriale DNA in der jüngeren Vergangenheit (vor ~ 170.000 Jahren) zu einem gemeinsamen Vorfahren zusammenläuft und dass die menschlichen Y-Chromosomen-Sequenzen ebenfalls zu einem gemeinsamen Vorfahren zusammenlaufen, sogar in noch jüngerer Vergangenheit (vor ~ 50.000 Jahren).48 Der scheinbare Konflikt ist, obwohl er vielfach in evolutionskritischer Literatur verwertet wird,49 ein Irrtum. Der Grund für das schnelle Zusammenlaufen der mitochondrialen und Y-Chromosomen-Sequenzen ist, dass diese DNA-Sequenzen in einer Weise vererbt werden, die sich von der von (nicht-Y-)chromosomaler DNA unterscheidet. Mitochondrien-DNA wird nur durch Mütter weitergegeben; Y-Chromosomen werden nur vom Vater an den Sohn weitergegeben. Von daher enden zurückverfolgte mitochondriale DNA-Abstammungslinien abrupt, wenn eine Mutter nur Söhne hat; in gleicher Weise enden Y-Chromosomen-Abstammungslinien schlagartig, wenn ein Vater nur Töchter hat. In beiden Fällen gehen nicht-Y-chromosomale DNA-Abstammungslinien jedoch weiter zurück (d. h. Väter und Mütter geben immer Chromosomen an Nachkommen beider Geschlechter weiter).
Betrachten wir eine Großfamilie (Abbildung 3). In diesem Beispiel erhalten alle Frauen in der dritten Generation ihre Mitochondrien-DNA von einem gemeinsamen weiblichen Vorfahren in der ersten Generation. Eine Untersuchung der Frauen in der dritten Generation würde zu folgenden Ergebnissen führen: Ihre mitochondriale Abstammungslinie würde schnell zusammenlaufen, aber ihre chromosomale DNA-Abstammungslinie nicht, da die Frauen ihre Chromosomen-DNA zum Teil (d.h. zu 50%) von zwei Personen in der zweiten Generation erhalten, die nicht mit der Quelle ihrer mitochondrialen DNA verwandt sind.
Dementsprechend weisen die Variationen in ihren genomischen Sequenzen darauf hin, dass sie einer größeren Population entstammen, die ihre mitochondriale DNA nicht bis in die Gegenwart weitergegeben hat. Mit anderen Worten, es wäre natürlich falsch zu schlussfolgern, dass ihr mütterlicher Vorfahre in der ersten Generation die einzige Frau war, die zu diesem Zeitpunkt gelebt hat oder zu behaupten, dass sie in einer Zeit eines massiven Bevölkerungsflaschenhalses lebte.
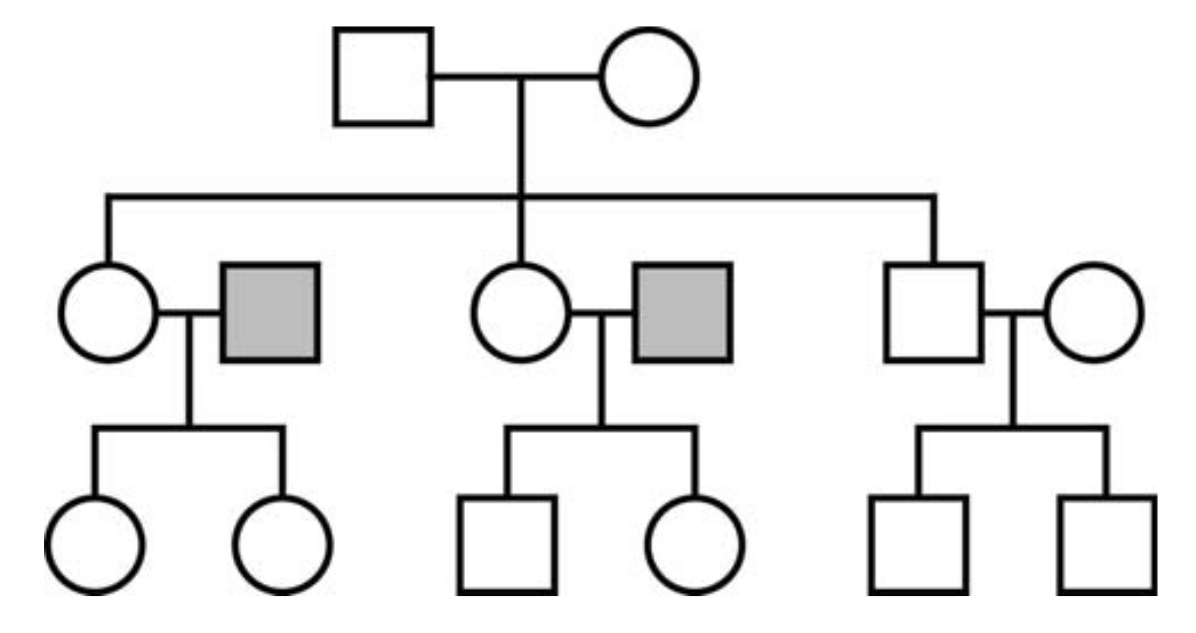
Abbildung 3. Mitochondriale und chromosomale Vererbung beim Menschen
Quadrate stehen für Männer, Kreise repräsentieren Frauen. Alle Frauen in der dritten Generation haben ihre mitochondriale DNA von ihrer gemeinsamen Großmutter geerbt; ihre chromosomale DNA haben sie jedoch auch von ihren Vätern (graue Quadrate) geerbt. Von daher sind Variationen in ihrer chromosomalen DNA die geeignete Grundlage zur Schätzung ihrer Populationsgröße.
Gleiches gilt für moderne menschliche Populationen. Obwohl unsere mitochondriale DNA-Abstammungslinie in relativ junger Vergangenheit in der »mitochondrialen Eva« zusammenläuft, zeigen heutige Variationen der menschlichen chromosomalen DNA, dass sie nur ein Mitglied einer umfangreichen sich fortpflanzenden Population war. Die gleiche Logik gilt entsprechend für die Vererbung des Y-Chromosoms und das Zusammenlaufen der Variationen des menschlichen Y-Chromosoms zu einem einzigen »Adam« in der jüngeren Vergangenheit. Obwohl das rasche Zusammenlaufen dieser speziell vererbten DNA-Sequenzen für sich genommen interessant ist, können solche Sequenzen wegen ihrer einzigartigen Vererbungsweise nicht herangezogen werden, um die Populationsgrößen bei unseren Vorfahren zu bestimmen.50
Genesis und das Genom: »Ratschen-Konkordismus« oder göttliche Akkomodation?
Die Erwartung, dass die Schöpfungserzählungen in der Genesis (erstes Buch Mose) naturwissenschaftlich gültige biologische Details über die menschliche Abstammung liefern, wird – so lässt sich zusammenfassen – angesichts der Erkenntnisse der Humangenomforschung in zweierlei Hinsicht völlig enttäuscht: Zum einen haben Menschen gemeinsame Vorfahren mit anderen Lebensformen; und zum zweiten geschah die Artbildung des Menschen durch eine sich untereinander mischende Population, nicht durch ein einziges Ahnenpaar. Von daher sind »naturwissenschaftlich konkordistische« Herangehensweisen an die Genesis durch diese Erkenntnisse jetzt stark unter Druck geraten.51 Die Erwartung, dass 1. Mose – mindestens zu einem gewissen Grad – naturwissenschaftlich gültige Informationen bietet, verbunden mit der Sichtweise, dass Naturwissenschaft ein berechtigtes Vorgehen ist, um ein zunehmend zuverlässigeres Verständnis der Schöpfungsordnung zu erhalten, erzeugt ein Phänomen, das ich als »Ratschen-Konkordismus« bezeichne. Dieser Ansatz ist daran erkennbar, dass diejenigen, die ihn verwenden, zunächst die Schlussfolgerungen neuerer Forschung, die zu ihren konkordistischen Erwartungen im Widerspruch stehen, ablehnen, indem sie Entscheidungen häufig mit der Behauptung, dass die Beweise nicht ausreichten, aufschieben. Wenn sich allerdings die Gegenbeweise zunehmend gegen ihre Position auftürmen, wird eine solche Person eventuell den betreffenden Punkt zugestehen, ihre spezielle konkordistische Erwartung verwerfen, und wie bei einer Ratsche zur nächsten verfügbaren Position überspringen, um das Gleichgewicht ihrer Erwartungen wieder herzustellen. In Anbetracht der hier vorgelegten Erkenntnisse der Genomforschung könnte zum Beispiel ein Positionswechsel von der Leugnung einer gemeinsamen Abstammung zu ihrer Annahme geschehen, wobei dann jedoch der Standpunkt beibehalten würde, dass die gemeinsame Abstammung biologisch von einem einzigen Paar in der jüngeren Vergangenheit ausgegangen wäre.52
Ein Rahmenkonzept »Schöpfung durch Evolution«, wie vor kurzem in den Werken von Denis Lamoureux vorgebracht,53 akzeptiert und integriert neue wissenschaftliche Erkenntnisse im Gegensatz zu einem »Ratschen-Konkordismus« bereitwillig. Dadurch, dass diese Sichtweise die »Naturwissenschaft« in den Schöpfungserzählungen der Genesis als göttliche Akkomodation (Anpassung) an die antike Kultur des Nahen Ostens behandelt, erwartet sie nicht, dass 1. Mose mit der modernen Naturwissenschaft übereinstimmt. Obwohl eine solche Auffassung als liberaler Umgang mit der Bibel kritisiert werden kann, trifft einen »Ratschen-Konkordismus« im Grunde die gleiche Kritik, da er postuliert, dass nur eine Teilmenge der Genesis zuverlässige naturwissenschaftliche Informationen enthält. Der konkordistische Ansatz geht zwar davon aus, dass die Genesis naturwissenschaftliche Informationen bereitstellen will, bestimmte naturwissenschaftliche Merkmale infolge einer Akkomodation aber ungenau oder verschleiert sind. Im Gegensatz dazu sieht der Ansatz »Schöpfung durch Evolution« die Schöpfungserzählungen als durchgängige Dokumente göttlicher Akkomodation an ihre ursprünglichen Hörer; als Erzählungen, die ohne die Absicht geschrieben wurden, moderne naturwissenschaftliche Belange anzusprechen.
 Dennis R. Venema ist außerordentlicher Professor und Leiter des Lehrstuhls Biologie an der Trinity Western University (Kanada). Er erwarb seinen BSc (Bachelor of Science) und seinen Doktortitel in Zellbiologie und Genetik an der University of British Columbia (Kanada). Seine Forschungsinteressen umfassen die Genetik von Gewebemusterbildung bei Drosophila, Genetikausbildung und das Zusammenspiel von Evolutionsbiologie und christlichem Glauben. Im Jahr 2008 wurde ihm der »College Biology Teaching Award« von der kanadischen Biologielehrervereinigung verliehen. Er ist Co-Autor einer Reihe von Blog-Beiträgen der BioLogos-Stiftung, in denen er vergleichende Genomforschung und die Evolution des Menschen erörtert. Zusammen mit seiner Frau Val hat er einen Sohn Elijah und eine Tochter Davin. Als Familie genießen sie zahlreiche Outdoor-Aktivitäten, die die Pazifikküstenregion in Kanada bietet.
Dennis R. Venema ist außerordentlicher Professor und Leiter des Lehrstuhls Biologie an der Trinity Western University (Kanada). Er erwarb seinen BSc (Bachelor of Science) und seinen Doktortitel in Zellbiologie und Genetik an der University of British Columbia (Kanada). Seine Forschungsinteressen umfassen die Genetik von Gewebemusterbildung bei Drosophila, Genetikausbildung und das Zusammenspiel von Evolutionsbiologie und christlichem Glauben. Im Jahr 2008 wurde ihm der »College Biology Teaching Award« von der kanadischen Biologielehrervereinigung verliehen. Er ist Co-Autor einer Reihe von Blog-Beiträgen der BioLogos-Stiftung, in denen er vergleichende Genomforschung und die Evolution des Menschen erörtert. Zusammen mit seiner Frau Val hat er einen Sohn Elijah und eine Tochter Davin. Als Familie genießen sie zahlreiche Outdoor-Aktivitäten, die die Pazifikküstenregion in Kanada bietet.
Originaltitel dieses Artikels: »Genesis and the Genome: Genomics Evidence for Human-Ape Common Ancestry and Ancestral Hominid Population Sizes«. In: Perspectives on Science and Christian Faith. Band 62, Nummer 3. September 2010.
Online verfügbar unter: http://www.asa3.org/ASA/PSCF/2010/PSCF9-10Venema.pdf
Deutsche Übersetzung: Kerstin und Mark Marzinzik (www.schoepfung-durch-evolution.de); Veröffentlichung mit freundlicher Genehmigung des Autors.
Artikel als PDF-Datei:
http://www.schoepfung-durch-evolution.de/media/Venema-Genesis-und-das-Genom.pdf
Das Titelbild dieses Artikels stammt aus folgender Quelle: https://commons.wikimedia.org/wiki/File:Human_male_karyotpe_high_resolution.jpg (Public Domain)
Anmerkungen und Literaturangaben
-
C. Darwin: The Descent of Man, and Selection in Relation to Sex. New York: D. Appleton and Company 1871. ↩
-
The Chimpanzee Sequencing and Analysis Consortium: Initial Sequence of the Chimpanzee Genome and Compari- son with the Human Genome. In: Nature 437. (2005). S. 69–87. ↩
-
International Human Genome Sequencing Consortium: Initial Sequencing and Analysis of the Human Genome. In: Nature 409 (2001). S. 860–920; International Human Genome Sequencing Consortium: Finishing the Euchromatic Sequence of the Human Genome. In: Nature 431. (2004). S. 931–45. ↩
-
The Chimpanzee Sequencing and Analysis Consortium; a.a.O. ↩
-
M. C. King und A. C. Wilson: Evolution at Two Levels in Humans and Chimpanzees. In: Science 188. (1975). S. 107–16. ↩
-
The Chimpanzee Sequencing and Analysis Consortium; a.a.O.; R. J. Britten: Divergence between Samples of Chimpanzee and Human DNA Sequences Is 5%, Counting Indels. In: Proceedings of the National Academy of Sciences of the USA 99. (2002). S. 13633–5. ↩
-
Ebda. ↩
-
R. Nielsen, C. Bustamante, A. G. Clark et al.: A Scan for Positively Selected Genes in the Genomes of Humans and Chimpanzees. In: PLoS Biology 3. (2005). S. 170. ↩
-
Die Insulin-Sequenzen der Tetrapoden in Abbildung 1 wurden zusammengestellt aus Daten, die über BLAST-Suchen von öffentlichen Genom-Datenbanken des National Center for Biotechnology Information (http://blast.ncbi.nlm.nih.gov/Blast.cgi?target=_blank) abgerufen wurden. ↩
-
Nielsen, Bustamante, Clark et al.; a.a.O. ↩
-
King und Wilson; a.a.O.; J. W. Ijdo, A. Baldini, D. C. Ward et al.: Origin of Human Chromosome 2: An Ancestral Telomere-Telomere Fusion. In: Proceedings of the National Academy of Sciences of the USA 88. (1991). S. 9051–5; T. Ried, N. Arnold, D. C. Ward und J. Wienberg: Comparative High-Resolution Mapping of Human and Primate Chromosomes by Fluorescence in Situ Hybridization. In: Genomics 18. (1993). S. 381–6. ↩
-
H. Kerher-Sawatzki, B. Schreiner, S. Tanzer et al.: Molecular Characterization of the Pericentric Inversion That Causes Differences between Chimpanzee Chromosome 19 and Human Chromosome 17. In: American Journal of Human Genetics 71. (2002). S. 375–88; vgl. auch Literaturverweise dort. ↩
-
L. W. Hillier, T. A. Graves, R. S. Fulton et al.: Generation and Annotation of the DNA Sequences of Human Chromosomes 2 and 4. In: Nature 434. (2005). S. 724–31; The Chimpanzee Sequencing and Analysis Consortium; a.a.O.; L. Feuk, J. R. MacDonald, T. Tang et al.: Discovery of Human Inversion Polymorphisms by Comparative Analysis of Human and Chimpanzee DNA Sequence Assemblies. In: PLoS Genetics 1. (2005). S. 56. ↩
-
Ijdo, Baldini, Ward et al.; a.a.O.; Hillier, Graves, Fulton et al.; a.a.O.; The Chimpanzee Sequencing and Analysis Consortium; a.a.O. ↩
-
Hillier, Graves, Fulton et al.; a.a.O. ↩
-
Obwohl die Mutationsrate innerhalb der Pseudogene größer erscheint als in funktionsfähigen Sequenzen (weil dort reinigende Selektion Mutationen aus der Population entfernt), ist sie tatsächlich in einem absoluten Sinn langsam wegen des Korrekturlesemechanismus durch DNA-Polymerasen während der Chromosomen-Replikation. ↩
-
D. Brawand, W. Wali und H. Kaessmann: Loss of Egg Yolk Genes in Mammals and the Origin of Lactation and Placentation. In: PLoS Biology 6. (2006). S. 0507–17. ↩
-
Ebda. ↩
-
Ebda. ↩
-
In diesem Artikel wird die Diskussion von Pseudogenen auf unitäre Pseudogene beschränkt: Sequenzen, denen eine homologe Sequenz innerhalb des gleichen Genoms fehlt, die aber im erwarteten syntenischen Abschnitt in funktionsfähiger Form in anderen Organismen vorhanden sind. Wenn man allerdings repetitive Elemente, Insertionen endogener Retroviren, prozessierte Pseudogene und so weiter betrachtet, könnten die Beispiele vervielfacht werden. ↩
-
T. Olender, D. Lancet und D. W. Nebert: Update on the Olfactory Receptor (OR) Gene Superfamily. In: Human Genomics 3. (2008). S. 87–97. ↩
-
Y. Gilad, O. Man, S. Pääbo and D. Lancet: Human Specific Loss of Olfactory Receptor Genes. In: Proceedings of the National Academy of Sciences of the USA 100. (2003). S. 3324–7. ↩
-
Ebda. ↩
-
M. Behe: The Edge of Evolution: The Search for the Limits of Darwinism. New York: Free Press 2007. ↩
-
Zum Beispiel versuchen zwei aktuelle populärwissenschaftliche ID-Bücher eine gemeinsame Abstammung von Mensch und Schimpanse in Zweifel zu ziehen und setzen gemeinsame Abstammung mit »Darwinismus« gleich: vgl. W. A. Dembski und S. McDowell: Understanding Intelligent Design: Everything You Need to Know in Plain Language. Eugene, OR: Harvest House 2008. S. 55–7 und C. Luskin und L. P. Gage: A Reply to Francis Collins’s Darwinian Arguments for Common Ancestry of Apes and Humans. In: Intelligent Design 101: Leading Experts Explain the Key Issues. Herausgegeben von H. W. House. Grand Rapids, MI: Kregel Publications 2008. S. 215–35. ↩
-
Dembski, McDowell; a.a.O. ↩
-
Luskin, Gage; a.a.O. ↩
-
Luskin, Gage; a.a.O. ↩
-
Drosophila 12 Genomes Consortium: Evolution of Genes and Genomes on the Drosophila Phylogeny. In: Nature 450. (2007). S. 203-18. ↩
-
S. W. Schaeffer, A. Bhutkar, B. F. McAllister et al.: Polytene Chromosomal Maps of 11 Drosophila Species: The Order of Genomic Scaffolds Inferred from Genetic and Physical Maps. In: Genetics 179. (2008). S. 1601–55; A. Bhutkar, S. W. Schaeffer, S. M. Russo et al.: Chromosomal Rearrangement Inferred from Comparisons of 12 Drosophila Genomes. In: Genetics 179. (2008). S. 1657–80. ↩
-
Ebda. ↩
-
Für ein Beispiel, das die wesentlichen ID-Ansätze zeigt, siehe: Luskin, Gage; S. 224–31. ↩
-
Für einen Überblick siehe N. A. Rosenberg und M. Nordborg: Genealogical Trees, Coalescent Theory and the Analysis of Genetic Polymorphisms. In: Nature Reviews Genetics 3. (2002). S. 380–90. ↩
-
A. Hobolth, O. F. Christensen, T. Mailund und M. H. Schierup: Genomic Relationships and Speciation Times of Human, Chimpanzee, and Gorilla Inferred from a Coalescent Hidden Markov Model. In: PLoS Genetics 3. (2007). S. 7. ↩
-
Zusätzliche Genbäume sind möglich, zum Beispiel wo Gendivergenz innerhalb einer Vorfahren-Population vor der Artbildung auftritt. Auch weitere Faktoren wie Hypermutabilität muss bei der Schätzung der Populationsgröße von diskordanten Gen-/Speziesbäumen berücksichtigt werden. Abbildung 2 ist zum Teil eine Anpassung und Verdichtung von Abbildung 1 in Holbolth, Christensen, Mailund und Schierup; a.a.O. Für eine eingehendere Erörterung dieser Sachverhalte siehe den ganzen Artikel; und Rosenberg und Nordborg; a.a.O. ↩
-
Rosenberg, Nordborg; a.a.O.; Hobolth, Christensen, Mailund und Schierup; a.a.O. ↩
-
Ebda. ↩
-
Ein Beispiel findet man in W. Li und L. A. Sadler: Low Nucleotide Diversity in Man. In: Genetics 129. (1991). S. 513–23. ↩
-
Hobolth, Christensen, Mailund und Schierup; a.a.O. ↩
-
F. C. Chen und W. H. Li: Genomic Divergences between Humans and Other Hominoids and the Effective Popula-tion Size of the Common Ancestor of Humans and Chimpanzees. In: American Journal of Human Genetics 68. (2001). S. 444–56; Z. H. Yang: Likelihood and Bayes Estimation of Ancestral Population Sizes in Hominoids Using Data from Multiple Loci. In: Genetics 162. (2002). S. 1811–23; Z. Zhao, L. Jin, Y. Fu et al.: Worldwide DNA Sequence Variation in a 10-kilobase Noncoding Region on Human Chromosome 22. In: Proceedings of the National Academy of Sciences of the USA 97 (2000). S. 11354–8. ↩
-
Hobolth, Christensen, Mailund und Schierup; a.a.O. ↩
-
Ebda. ↩
-
Siehe www.hapmap.org ↩
-
A. Tenesa, P. Navarro, B. J. Hayes et al.: Recent Human Effective Population Size Estimated from Linkage Disequi- librium. In: Genome Research 17. (2007). S. 520–6. ↩
-
Ebda. ↩
-
Ebda. ↩
-
Die christliche Organisation Reasons to Believe hält an einem buchstäblichen Adam und einer buchstäblichen Eva als Vorfahren der gesamten Menschheit fest und datiert sie auf etwa 50.000 Jahre vor heute. ↩
-
M. Ingman, H. Kaessmann, S. Pääbo und U. Gyllensten: Mitochondrial Genome Variation and the Origin of Modern Humans. In: Nature 408 (2000). S. 708–13; R. Thomson, J. K. Pritchard, P. Shen et al.: Recent Common Ancestry of Human Y Chromosomes: Evidence from DNA Sequence Data. In: Proceedings of the National Academy of Sciences of the USA 97 (2000). S. 7360–5. ↩
-
Siehe zum Beispiel F. Rana und H. Ross: Who Was Adam? (Colorado Springs: Navpress, 2005). S. 123–31. ↩
-
F. Ayala, A. Escalante, C. O’Huigin und J. Klein: Molecular Genetics of Speciation and Human Origins. In: Proceedings of the National Academy of Sciences of the USA 91. (1994). S. 6787–94. ↩
-
D. Lamoureux: Evolutionary Creation: A Christian Approach to Evolution. (Eugene, OR: Wipf & Stock, 2008). ↩
-
Denis Lamoureux (persönliche Mitteilung) hat auch anekdotische Belege von Personen gesammelt, die im Lichte neuer Erkenntnisse von einer konkordistischen Position zur nächsten »ratschen«. Seiner Erfahrung nach sind die bedeutendsten »Positionen« auf der Ratsche: »Junge-Erde-Kreationismus«; »Alte-Erde-Kreationismus«; »Schöpfung durch Evolution«, wobei an einem buchstäblichen Adam und einer buchstäblichen Eva als biologische Vorfahren der Menschheit festgehalten wird (evolutionärer Monogenismus); und letztlich echte »Schöpfung durch Evolution« (ohne verbleibende naturwissenschaftlich-konkordistische Erwartungen an die Genesis). Weitere Abstufungen sind natürlich möglich. ↩
-
D. Lamoureux: Evolutionary Creation: A Christian Approach to Evolution; a.a.O.; D. Lamoureux: Lessons From the Heavens: On Scripture, Science and Inerrancy. In: Perspectives on Science and Christian Faith 60. (2008). S. 4–15. ↩